


What do you want to do?
GENERATE INPUT STRUCTURES
Here is the relevant section of the tutorial.
Modify the gen_conformers.sh according to your topology and desired number of structures, and run it.
Then, modify and run the prune_conformers.sh script to remove unreasonable structures. The resulting structure set will be in TRAJECTORY format.
CREATE QUANTUM INPUT FILES
Here is the relevant section of the tutorial.
You must set the following options in the job control file:
- RUNTYPE=CREATE_INPUT
- NSTRUCTURES=[number of structures]
- COORDINATE_FORMAT=[TRAJECTORY/RESTART]
- QMFILEFORMAT=[ADF/GAMESS/GAUSSIAN]
- QMHEADER=[header file]
- QMFILEOUTSTART=[file prefix]
- QMFILEOUTEND=[file suffix]
- QM header file to be prepended to all QM input files
- Topology file
- Coordinate file
paramfit -i [job control] -p [topology] -c [coordinates]
SET PARAMETERS TO FIT
You must set the following options in the job control file:
- RUNTYPE=SET_PARAMS
- PARAMETER_FILE_NAME=[filename]
- Topology file
paramfit -i [job control] -p [topology]
DERIVE K
You must set the following options in your job control file:
- RUNTYPE=FIT
- PARAMETERS_TO_FIT=K_ONLY
- FUNC_TO_FIT=SUM_SQUARES_AMBER_STANDARD
- NSTRUCTURES=[number of structures]
- COORDINATE_FORMAT=[TRAJECTORY/RESTART]
- QM_ENERGY_UNITS=[HARTREE/KCALMOL/KJMOL]
paramfit -i [job control] -p [topology] -c [coordinates] -q [quantum energy file]
PERFORM A SINGLE MOLECULE FIT TO ENERGIES
Using the wizard to generate the job control file may be simpler than creating one by hand. However, if you wish to do so, you must set the following options in your job control file:
- RUNTYPE=FIT
- COORDINATE_FORMAT=[TRAJECTORY/RESTART]
- NSTRUCTURES=[number of structures]
- K=[derived K value]
- FUNC_TO_FIT=SUM_SQUARES_AMBER_STANDARD
- QM_ENERGY_UNITS=[HARTREE/KCALMOL/KJMOL]
- PARAMETERS_TO_FIT=[LOAD/DEFAULT]
- PARAMETER_FILE_NAME=[parameter file, if loading]
- ALGORITHM=[GENETIC/SIMPLEX/BOTH/NONE]
- Additional algorithm parameters, if desired
- Output options, if desired
paramfit -i [job control] -p [topology] -c [coordinates] -q [quantum energy file]
DIAGNOSE A BAD FIT
Here is the relevant section of the tutorial.
First, determine that you do in fact have a bad fit. Are the resulting parameters unreasonable? Is the final function value very high, or the R2 very low? If so, troubleshoot with the following steps:
- Are your units correct? Check the value of QM_ENERGY_UNITS in your job control file, and read the documentation for your quantum package to make sure that Paramfit knows what units the quantum energies are in and can successfully convert them to kcal/mol, which it uses internally.
- Is K set properly? Did you derive K with the exact same input structures, QM data, and weights with which you performed the fit? Set WRITE_ENERGY=[filename], redo the fit, and plot.x [filename] to visualize the energy profile. If the Amber+K energy is consistently different by a constant factor, adjust K accordingly.
- Is the algorithm prematurely converging? Run it again, with exactly the same inputs,
outputs, and job control file. Are the results different each time? If so, the algorithm isn't converging
on the global minimum.
Premature convergence results from poor choice of algorithm settings, or more likely from providing Paramfit with too few input structures relative to the number of parameters you want to fit. To fix, adjust the algorithm parameters and/or provide more input structures. - Do the input structures provide adequate sampling? Do the input structures adequately sample the range of possible values? Check by setting SCATTERPLOTS=ON in the job control file, looking for warnings from the bounds checking functionality, and visualizing the resulting plots with scatterplots.sh in Paramfit's src directory.
- Are high-energy structures biasing the fit? If sampling is okay, check that some structures aren't biasing the fit. Set WRITE_ENERGY=[filename], redo the fit, then plot.x [filename] to examine the energy profile. Weight structures that bias the fit less, or ignore them altogether.
CHANGE WEIGHTING OF DIFFERENT STRUCTURES
Append a column to your QM energy file (separated by a space). This column contains the weight of each structure. Any structures that do not have a value in this second column will be assigned the default weight of 1. Weights smaller than this mean a structure is less important in the fit. Larger, and they are more important.
The fitness function with weighting is:
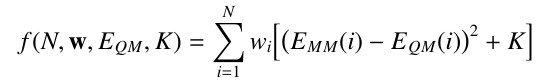
For example, the following QM energy file would make structure 2 twice as important to the fit as structures 1 and 3, while structure 4 would be half as important:
112.0 98.3 2.0 113.0 212.5 0.5
SETUP MULTIPLE MOLECULE FIT
TWEAK FITTING ALGORITHM PARAMETERS
If you have reason to believe the fitting algorithm is prematurely converging (for example, if running Paramfit multiple times produces a poor fitness function value and different parameters each time), you can adjust the various algorithm parameters.
Paramfit includes two fitting algorithms: a hybrid genetic algorithm that incorporates simplex refinements, and a simplex algorithm. The genetic algorithm excels at finding the neighborhood of the global minimum, while its simplex refinements find the global minimum within this neighborhood. The simplex algorithm is analogous to an amoeba that crawls around the fitness function surface in search of the lowest point, and has a tendency to converge prematurely in local minima, which are numerous in this problem. This algorithm is highly dependent on the initial parameters, and should only be used for small refinements. If in doubt, use the hybrid genetic algorithm, as it incorporates the best traits of the simplex algorithm as well. For more details on the algorithms Paramfit uses, please read the paper.
The following table lists the adjustable parameters of the genetic algorithm. Most likely you will not need to adjust these values, and if so only change one or two at a time.
| Job Control Entry | Default | Recommended range | Function |
|---|---|---|---|
| OPTIMIZATIONS | 50 | 20-100 | The population size. Higher provides more genetic diversity, but is slower. |
| MAX_GENERATIONS | 10000 | >5000 | The maximum number of generations before the algorithm is killed. Too small, and the algorithm may prematurely converge. |
| GENERATIONS_TO_CONV | 20 | 5-50 | Number of generations that must pass, in a row, without any improvement in the fitness of the best parameter set in the population. Larger values establish a stricter convergence criterion, but will make the algorithm take longer. |
| GENERATIONS_TO_SIMPLEX | 5 | 3-5 | Number of generations that must pass without improvement to trigger a simplex refinement on 20% of the population. Set to 0 for no simplex refinement (will result in much slower convergence). Too small and the algorithm may converge to a local minimum, too large and it will take a large number of generations to converge. |
| GENERATIONS_WITHOUT_SIMPLEX | 10 | 5-10 | If GENERATIONS_TO_SIMPLEX != 0, number of generations that must pass between calls to simplex refinement. Too small and the genetic algorithm will not have time to incorporate refined values into the population, leading to convergence at local minima. Too large and it will take a large number of generations to converge. |
| MUTATION_RATE | 0.10 | 0.05-0.20 | The percentage of parameter values in the population that can be mutated each generation. Too high and good parameter sets will be destroyed, too low and convergence will take a long time |
| PARENT_PERCENT | 0.25 | 0.20-0.30 | The percentage of the population that is allowed to pass on parameters to the next generation. Too high and bad parameters will not be selected out, too low and it may fall into local minima. |
| SEARCH_SPACE | -1 | depends | The spread of values in the initial population. Set to -1 to search all of valid parameter space, or between 0 and 1 to search around that percentage of the given parameters |
GET THE PARAMFIT PAPER
CITE PARAMFIT
Betz, R.M.; Walker, R.C.; "Paramfit: Automated optimization of force field parameters for molecular dynamics simulations." Manuscript submitted for publication.